3月10日,蘭大二院神經內科二病區在王滿俠教授的指導下成功為一名17歲男性脊髓性肌萎縮癥(Spinal muscle atrophy,SMA)3型患者順利實施了鞘內注射“諾西那生鈉” Spinraza(nusinersen)。副主任醫師李鑫在治療前通過脊肌萎縮癥上肢模塊修訂版(RULM)、6分鐘步行試驗(6MWT)等量表對患者進行了個體化的精準評估,主治醫生王力群、栗靜協同完成藥物鞘內注射。目前,在醫院各部門的支持與協調下,蘭大二院已在小兒神經科開展了SMA-1型及2型患兒治療,在神經內科二病區開展了SMA-3型患者靶向藥物治療。

SMA致病基因是位于5q13的存活運動神經元1 (survival motor neuron 1, SMN1)基因,多數患者是因SMN1基因exon 7(可伴exon 8)純合缺失而致病,有95%靈敏度和100%特異性,所以又稱5q-SMA。目前SMA的確診主要依靠基因檢測。諾西那生鈉注射液是一種反義寡核苷酸(ASO),通過鞘內注射給藥,用于治療5q-SMA,臨床研究發現,諾西那生鈉鞘內注射治療可顯著提高SMA1型患者生存率,改善吞咽、呼吸功能,持續達到運動里程碑;持續改善和穩定SMA2、3型患者的運動功能;穩定SMA患者的病情并改善運動功能,改變自然病程。諾西那生鈉注射液作為全球首個SMA精準靶向治療藥物,2019年進入中國市場時價格較為昂貴,臨床應用受到極大限制,2021年12月國家醫保局將其納入新版醫保藥品目錄,經過醫保報銷,患者只需自費約1萬元即可注射一針,這對SMA患者及其家庭帶來了希望。
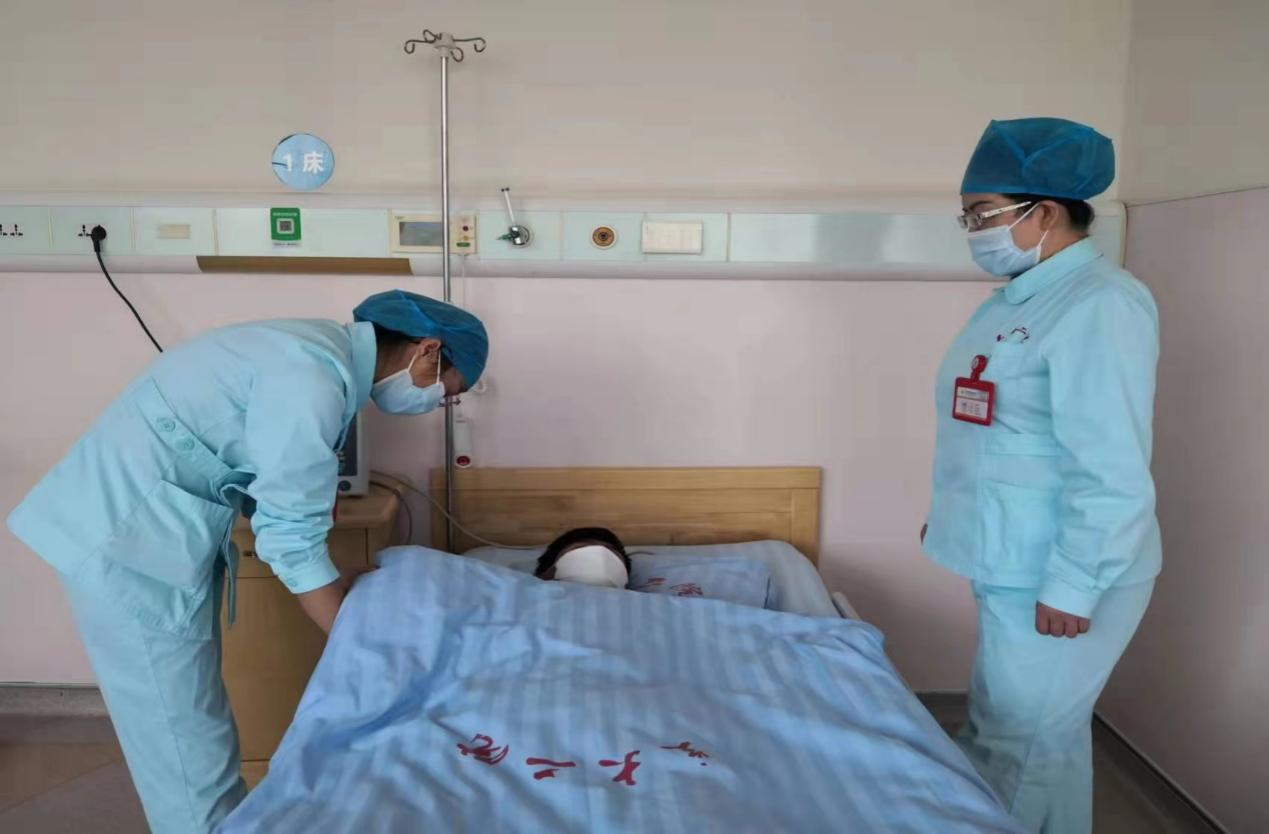
目前,神經內科二病區已確診3例患者,“諾西那生鈉”治療能幫助SMA患者生活開啟新篇章。針對治療,神經內科二病區主任醫師武國德強調:“在注射前要對于脊髓性肌萎縮癥患者進行個體化評估,對嬰幼兒患者使用Hammersmith神經檢查模塊、CHOP INTEND以及WHO Moter Milestones等量表評估,成人患者使用脊肌萎縮癥上肢模塊修訂版及6分鐘步行試驗等,同時在整體治療過程中動態監測患者運動功能等改善情況,這樣才能最大化地提高患者生存率,持續改善吞咽、呼吸及運動功能,隨著諾西鈉生鈉注射液、利司撲蘭口服溶液等針對脊髓性肌萎縮癥的藥物不斷用于臨床治療,相信會有更多的患者受益。”
神經內科二病區作為國家神經系統疾病臨床醫學研究中心-神經免疫與感染疾病研究分中心、甘肅省重點學科、甘肅省神經病學疾病精準診療技術國際合作基地、甘肅省神經內科專業醫療質量控制中心、蘭州大學臨床醫學研究型學科、蘭州大學第二醫院藥物臨床試驗機構神經內科專業基地,科室全體醫務人員始終堅持以患者為中心,以提高醫療質量為宗旨,愛崗敬業,開拓進取,竭誠為廣大神經科患者提供優質服務。
【科普常識】
脊髓性肌萎縮癥(Spinal muscle atrophy,SMA)是一種常染色體隱性遺傳的神經肌肉疾病,以脊髓前角或腦干運動核α運動神經元變性和丟失為特征,主要臨床特點為進行性、對稱性四肢和軀干的肌無力,近端重于遠端,下肢重于上肢,有時可見舌肌纖顫、手震顫。SMA患者起病年齡差異性大,從出生前至成人期均可發病,發病率為1/10000~1/6000,攜帶者頻率為1/40-1/60,我國約為1/46。中國現有SMA患者2萬~3萬人,每年新發病例大概850人。SMA根據發病年齡以及最大運動功能可分為不同亞型,約80%為1、2型,是兒科神經肌肉病常見的一種遺傳病。SMA是嬰幼兒致死性遺傳疾病之一,發病越早,壽命越短,預后越差,如果得不到有效治療,多數嬰兒型患兒在0~2歲內死于呼吸衰竭。3型或4型SMA患者發病隱匿,診斷較困難,需臨床特征、實驗室輔助檢查結合基因檢測而確診。
相關新聞
- 2017-01-20隴周刊(2017年 第3期)
- 2017-01-26隴周刊(2017年 第4期)
- 2017-02-10 隴周刊(2017年 第5期)
- 2017-02-17 隴周刊(2017年 第6期)
精彩推薦
-
 蘭州永登:新鮮蘑菇上市 鎮黨委書記幫忙找銷路
蘭州永登:新鮮蘑菇上市 鎮黨委書記幫忙找銷路 -
 【小康路上看老鄉】文縣:6000多畝蒜薹喜獲豐收
【小康路上看老鄉】文縣:6000多畝蒜薹喜獲豐收 -
 蘭州市城關區:他們奮戰抗疫一線 每位平凡的身影都值得點贊
蘭州市城關區:他們奮戰抗疫一線 每位平凡的身影都值得點贊 -
 【暢游隴原】沙漠深處的摘星小鎮
【暢游隴原】沙漠深處的摘星小鎮 -
 加強保護傳承利用 打造中華文化標志——《長城國家文化公園(甘肅段)建設保護規劃》解讀
加強保護傳承利用 打造中華文化標志——《長城國家文化公園(甘肅段)建設保護規劃》解讀 -
 甘肅省第十五屆運動會志愿者開始面試選拔
甘肅省第十五屆運動會志愿者開始面試選拔 -
 蘭州市城關區消費者協會發布“3·15”維權典型案例
蘭州市城關區消費者協會發布“3·15”維權典型案例 -
 甘肅省從嚴監管特種設備 保護群眾生命財產安全
甘肅省從嚴監管特種設備 保護群眾生命財產安全